Bu yazı da ise R programlama kullanarak ScRNA-seq veri setini çeşitli ön işlemelerden geçirdikten sonra denetimsiz (unsupervised) makine öğrenmesi yöntemlerinden olan kümeleme (clustering) algoritmalarını kullanarak veri setini kümerlere ayıracağız. İsterseniz Python programlama diliyle de analiz yapabilirsiniz ama ben stajda R kullandığım için bu yazıda da R kullanacağım. Yazının sonuna Python kullanarak gerçekleştirebileceğiniz eğitici linkler de koyacağım. Haydi başlayalım!!
Gerekli Kurulumlar:
Basitliği açısından analizi dropClust adlı R paketi ile yapacağız. R paketinin geliştirici sürümü, aşağıdaki R komutlarıyla kurulabilir:
library(devtools)
install_github(“debsin/dropClust”, dependencies = T)
Bu yazıda, standart bir pipeline göstermek için 10X web sitesinden (3K PBMC veri seti) küçük bir veri seti kullanacağız. Burada göstereceklerim bu veri setinin en basit haliyle analiz edilmesi olarak düşünebilirsiniz. Yazının sonunda kendini daha çok geliştirmek isteyenler için bazı linkler vereceğim ve ayrıca benin staj zamanı yaptığım projenin Github linkini de koyacağım.
Gerekli Kütüphaneyi Çağırma:
library(dropClust)
set.seed(0)
Veri Seti Yükleme:
İndirdiğimiz veri setinin içinde 3 çeşit özellik vardır. Bunlar:
- Count Matrix
- Transkriptome tanımlayıcıları
- Gen tanımlayıcıları
# Load Data
sce <-readfiles(path = “C:/Projects/dropClust/data/pbmc3k/hg19/”)
Veri Ön işleme (Düşük Kaliteli Gen ve Hücrelerin Filtrelenmesi):
dropClust, düşük kaliteli hücreleri ve genleri çıkarmak için ön işleme gerçekleştirir. dropClust ayrıca mevcut olabilecek toplu etkileri (batch effect) azaltmak için donatılmıştır. Kullanıcının, bireysel transkriptomlar için serinin kaynağına ilişkin herhangi bir bilgi sağlaması gerekmez. Ancak, toplu etki kaldırma adımı isteğe bağlıdır.
Hücreler, min_count parametresi tarafından belirtilen bir hücredeki toplam UMI sayısına göre filtrelenir. Düşük kaliteli genler, belirli bir min_count eşiğinin üzerindeki ifadelerle min_count minimum hücre sayısına göre çıkarılır.
# Filter poor quality cells. A threshold th corresponds to the total count of a cell.
sce<-FilterCells(sce)
sce<-FilterGenes(sce)
Ayrıca şunu belitmek istiyorum ki bu yazıda kullanılan bütün dropClust fonksiyon parametreleri bu veri setini en iyi şekilde analiz edecek şekilde default değerleri almaktadır. İsterseniz ?(fonksiyon_adi) komutunu kullanarak parametreler hakkında bilgi alıp bu parametreleri veri setinize göre daha iyi bir sonuç elde etmek için tune edebilirsiniz.
Veri Normalizasyonu
Count normalizasyonu daha sonra sadece kaliteli genlerle üzerinden gerçekleştirilir. Normalleştirmenin amacı, istenmeyen teknik varyasyonu ortadan kaldırmaktır. Normalleştirilmiş ifade değerleri, medyan normalleştirilmiş toplam sayı kullanılarak bir SingleCellExperiment nesnesindeki ham count verilerinde hesaplanır (SingleCellExperiment nesnesi apayrı bir konu. Eğer burada bahsedersem yazımız çok uzayacak. Bu linkten daha detaylı bilgiler öğrenebilirsiniz). Normalizasyon işlemi aşağıdaki fonksiyon ile gerçekleştirilir.
sce<-CountNormalize(sce)
Gen Seçimi:
Daha fazla gen seçimi, genlerin dağılım indeksine göre sıralanmasıyla gerçekleştirilir.
# Select Top Dispersed Genes by setting ngenes_keep.
sce<-RankGenes(sce, ngenes_keep = 1000)
Örnekleme:
Birincil kümeleme, verilerin brüt yapısını tahmin etmek için hızlı bir şekilde gerçekleştirilir. Bu kümelerin her biri daha sonra kümeleme işlemine ince ayar yapmak için örneklenir.
sce<-Sampling(sce)
Boyutsal İndirgeme:
RNAseq verileri yüksek boyutlu verilerdir. Bu sebeple, PCA gibi çeşitli yöntemler kullanarak bu veri setinin boyutunu indirgememiz gerekmektedir. PCA, ana bileşenleri etkileyen genleri tanımlamak için kullanılır.
sce<-RankPCAGenes(sce)
Kümeleme:
Son olarak veri setini çeşitli algoritmalar kullanarak kümelere ayırmalı ve görselleştirmeliyiz. Varsayılan olarak en uygun, Louvain tabanlı kümeler döndürülür. Ancak kullanıcı, istenen sayıda kümeyi üretmek için parametreleri ayarlayabilir. Örneklenmemiş transkriptomlara, ince ayar kümelemesinden üretilen tanımlayıcılar arasından küme tanımlayıcıları atanır. Post-hoc atama, conf güven değeri ayarlanarak kontrol edilebilir. Yüksek conf değerleri, yalnızca ortak en yakın komşuların çoğunluğunu paylaşan transkriptomlara küme tanımlayıcıları atayacaktır.
# When `method = hclust`
# Adjust Minimum cluster size with argument minClusterSize (default = 20)
# Adjust tree cut with argument level deepSplit (default = 3), higher value produces more clusters.
sce<-Cluster(sce, method = “default”, conf = 0.8)
Kümeleri Görselleştirme:
sce<-PlotEmbedding(sce, embedding = “umap”, spread = 10, min_dist = 0.1)plot_data = data.frame(“Y1” = reducedDim(sce,”umap”)[,1], Y2 = reducedDim(sce, “umap”)[,2], color = sce$ClusterIDs)ScatterPlot(plot_data,title = “Clusters”)

Hangi genlerin hangi kümelerde yoğunluk gösterdiğini bulmak için de aşağıdaki kodu kullanıyoruz.
DE_genes_all = FindMarkers(sce, selected_clusters=NA, lfc_th = 1, q_th =0.001, nDE=30)
write.csv(DE_genes_all$genes,
file = file.path(tempdir(),"ct_genes.csv"),
quote = FALSE)marker_genes = c("S100A8", "GNLY", "PF4")
p<-PlotMarkers(sce, marker_genes)
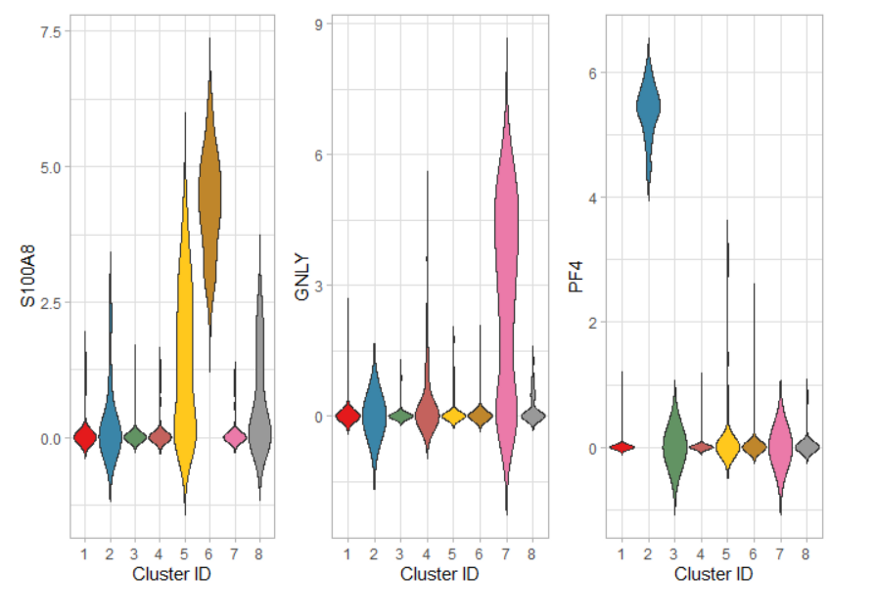
Kümeleme sonuçlarımızı yorumlamak için kümeler arasında ayrılmayı sağlayan genleri tanımlarız. Bu işaretleyici genler, işlevsel açıklamalarına dayalı olarak her kümeye biyolojik anlam atamamızı sağlar. En belirgin durumda, her küme için işaretleyici genler, kümelemeyi hücre tipi kimliği için bir vekil olarak ele almamıza izin vererek, belirli hücre tipleri ile önsel olarak ilişkilidir. Aynı ilke, kümeler arasındaki daha ince farkları keşfetmek için de uygulanabilir.
Bu yazıda Tek Hücreli RNA Dizileme Verilerinin en basit haliyle dropClust kütüphanesini kullanarak veri analizini yaptık. Umarım beğenmişsinizdir. Bu yazının devamında daha detaylı bilgiler verip kütüphane kullanmadan, veri ön işleme ve kümeleme süreçlerinin arkasındaki matematiği öğreterek bu işlemleri gerçekleştirmek istiyorum.
Buraya kadar sabredip okuduğunuz için teşekkür ederim.
KAYNAK -> Turhan Can Kargın Medium Hesabı
